肿瘤干细胞是致命肿瘤的源泉之一。而根据斯坦福大学完成的一项最新研究,维生素C+抗生素组合可以清除肿瘤干细胞。
根据这项发表在Oncotarget上的新研究,这种抗生素叫做多西环素如果在维生素C作用后加入,可以在实验室条件中高度有效地杀死肿瘤干细胞。
研究人员解释说这种方法提供了一种预防癌细胞耐药的新方法,展示了如何采用联合治疗克服肿瘤细胞耐药性。
Michael Lisanti教授设计了这项实验,他解释说:"我们现在知道一部分肿瘤细胞可以躲过化疗药物的杀伤,从而变成耐药细胞,而我们的这种新策略揭示了这个过程是如何发生的。"
"我们怀疑问题的答案在于某些肿瘤细胞可以变更它们的能量来源。因此当药物治疗减少某些能量来源时,这些肿瘤细胞可以通过其他的能量来源维持生存。"
这种新的联合方式防止了肿瘤细胞改变它们的能量来源,通过防止它们从其他物质获得能量,从而有效地让这些细胞饥饿。
这个来自斯坦福大学生物医学研究中心的研究团队发现采用不断增加的多西环素处理三个月可以使癌细胞变成代谢不灵活的细胞。结果就是癌细胞可以存活,但是却被严重抑制,所以它们对饥饿异常敏感。
研究人员首先通过限制肿瘤细胞仅仅采用葡萄糖为唯一能量来源,从而抑制了肿瘤细胞的线粒体,随后他们去掉了葡萄糖,进而有效饥饿癌细胞至死。
"在这种情况下,维生素C发挥糖酵解抑制剂的功能,该过程是线粒体供能的过程,而线粒体是细胞的能量源。"论文共同作者Federica Sotgia博士解释道。
该团队最近发现维生素C抑制肿瘤细胞生长的疗效比2-DG等药物高10倍,但是他们解释说当维生素C与抗生素联合使用时,其疗效会增加10倍,使得其抑癌效果是2-DG的100倍之多。
由于多西环素和维生素C都是无毒的,因此这种方法可以显著降低抗癌疗法的副作用。
该团队还发现其他药物也可以作为抗生素处理后的第二剂炸弹,如FDA批准的廉价无毒药物黄连素。
Lisanti教授补充道:"还需要进一步的证据证明维生素C和其他无毒药物也许在抗癌中发挥重要作用。我们的结果表明它是一种很有潜力的临床试验药物,通过将之添加到许多传统的化疗方案中,可以进一步防止肿瘤复发、进展和转移。"
近日,一项刊登在国际杂志Science上的研究报告中,来自耶鲁大学的研究人员开发了一种新方法,未来这种新方法或有望帮助开发出新型的抗生素。如今多种类型的细菌都开始对当前抗生素产生耐受性,这无疑就会增加致死性感染发生的频率,而新型抗生素的开发对于感染性疾病患者的治疗和公众健康的保护都显得非常重要。
研究者Seth Herzon教授说道,作为合成化学家,开发新型抗生素是唯一能够帮助治疗患者的方法,本文中我们所开发的新方法能够在实验室中通过简单的商业化学品来制造和截短侧耳素类抗生素(pleuromutilin)相关的分子,截短侧耳素类抗生素是真菌所产生的,早在20世纪50年代研究人员就发现这种类型的抗生素具有抗菌特性,从那时开始,学术界和制药领域的科学家们就通过半合成的方法开发出了数千种截短侧耳素衍生物,由于半合成的方法仅会对截短侧耳素本身进行化学性修饰,因此大部分的衍生物仅会在分子的单一位置发生改变,而完全合成方法则能够开发出额外的新型抗生素,但目前研究人员仍然并不清楚如何进行完全的合成。
2008年,研究者Herzon通过首次尝试发现了一种溶液,他表示,我们从事这项研究计划已经很多年了,但似乎并未获得很多成功;从历史上来看,制药行业一直是抗生素开发背后的强大驱动力,然而从投资回报角度来看,抗生素的开发似乎排名较靠后,而且大多数制药公司已经离开了新型抗生素研发的领域了,因此这就引发了抵御耐药性新型药物的严重匮乏,研究者补充道,随着抗菌危机不断恶化,我们就需要重新思考使用一种新型的策略来帮助开发抗生素。
如今研究人员能够制造出抗生素截短侧耳素的同分异构体,这种化合物具有相同的连接性,但分子排列不同,而且在合成截短侧耳素的最后步骤还能对其进行重排;本文研究或能帮助研究人员突破此前研究中的障碍对抗生素截短侧耳素进行完全合成,此外,相比截短侧耳素自身而言,其同分异构体具有更好的抗菌特性,这就为研究者后期开发改进型的化合物奠定了基础。
最后研究者Herzon说道,能够开发出抗生素截短侧耳素是非常伟大的,但我们更感兴趣的是我们可以通过合成的方法开发出非天然性的化合物,后期我们将继续优化合成过程,同时开始对开发出的新型化合物进行检测,如果一切顺利,研究者希望将所开发出的新型化合物推向临床试验来检测其在药物耐受性感染治疗上的效果。
最近,一项发表于国际著名杂志Cell上的研究报告中,来自MIT和哈佛大学的研究人员通过研究发现,肠球菌(enterococci)作为院内感染的主要"超级细菌"或许产生自距今4.5亿年前的祖先,而那时候动物刚刚从海洋爬行到陆地生活,也就是说,这个时间还要早于恐龙时代,文章中,研究者阐明了肠球菌(属)的进化历史,同时研究者还发现,这种细菌进化出了坚不可摧的特性,而且其也是如今引发医院内抗生素耐药感染的主要原因。
抗生素耐药性是引发全球人群健康的重大隐患,被认为是超级细菌的微生物往往对目前几乎所有抗生素都能够产生耐药性,当然这也是医院非常重视的问题,目前全球的科学家都在寻找能够解决抗生素耐药性的方法,因此,理解抗生素耐药性的进化或许就能够给研究者们带来一定帮助。
研究者Ashlee M. Earl博士表示,通过分析当今肠球菌的基因组和行为,我们就能够将时钟调回至这种细菌最初存在的形式,同时还能够绘制出肠球菌不断变化的图谱,理解肠球菌在环境中产生"特性"的分子机制或许能够帮助研究人员预测这种细菌适应抗生素及抗菌肥皂的方式,也为开发有效控制这种细菌扩散的方法提供了新的思路。
文章中,研究者发现,包括那些从未在医院发现的肠球菌,所有的肠球菌属细菌都能够对干燥、饥饿、消毒剂以及很多抗生素产生天然耐受性,正常情况下肠球菌生活在大部分陆地动物的肠道中,当然其也会生活在如今已经灭绝的动物肠道中,比如恐龙以及首个爬行到陆地生活的千足虫样的有机体;对肠球菌基因组的对比分析就能够为科学家们提供研究证据,实际上,研究人员还发现,每当新型动物物种出现时就会出现新型的肠球菌,比如当首次爬行到陆地后动物新物种的出现,以及物种大量灭亡后新物种的产生等,尤其是大约2.5亿年前发生的二迭纪末大灭绝事件。
在诸如鱼类的海洋动物中,肠道微生物都会被排入海洋中,每滴水中大约含有5000种无害的细菌,它们会沉到富含微生物的沉淀物中,而且通常会被蠕虫、贝类以及其它海洋中的食腐动物所消化,随后这些动物再被鱼类吃掉,因此这些微生物就会在食物链中不断循环,然而,在陆地上,肠道微生物往往会以粪便的形式被排出,随着时间延续这些微生物就会死亡。然而肠球菌通常比较耐干燥以及饥饿,其能够很好地在环境中生存。
Michael S. Gilmore博士表示,目前我们非常清楚经过数百万年的进化后肠球菌所携带的特殊基因,当然这些基因也驱动肠球菌能够抵御多种残酷的环境,但我们可以利用这些基因作为新型靶点来帮助开发新型的抗生素以及消毒剂有效地清除掉肠球菌,从而有效抵御肠球菌引发的院内耐药性感染。
在一项新的研究中,研究人员报道他们如今知道如何构建一种能够穿透革兰氏阴性菌的分子特洛伊木马,从而解决了一个几十年来一直阻止着为越来越有耐药性的细菌开发有效的新的抗生素的问题。相关研究结果于2017年5月10日在线发表在Nature期刊上,论文标题为"Predictive compound accumulation rules yield a broad-spectrum antibiotic"。
在美国伊利诺伊大学化学教授Paul Hergenrother的领导下,这些研究人员通过修饰一种仅杀死革兰氏阳性菌的药物来测试了他们的方法。革兰氏阳性菌缺乏坚固的外膜,但是革兰氏阴性菌的一大特征就是含有这种坚固的外膜,这就使得很难抵抗它们。他们报道,这种修饰将这种药物转化为一种广谱抗生素,从而也能够杀死革兰氏阴性菌。
革兰氏阴性菌包括大肠杆菌的致病性菌株、不动杆菌、克雷伯氏菌和铜绿假单胞菌,根据美国疾病控制与预防中心(CDC)的说法,这些细菌"对大多数可用的抗生素正在变得越来越有耐药性"。
Hergenrother说,发现新的抗生素来抵抗这些病原菌的努力一次又一次地失败了,原因仅是几乎所有的新药物都不能够穿透革兰氏阴性菌的细胞壁。
Hergenrother说,"我们已有一些抵抗革兰氏阴性菌的抗生素类型,但是最后一类抗生素是在50年前(即1968年)开发出来的。如今,这些细菌正在对所有的这些抗生素产生耐药性。"
没有新的抗生素出现并不是因为缺乏努力。比如,在2007年,一家大型制药公司筛选了大约50万种化合物是否具有抵抗大肠杆菌的活性,结果没有一种化合物成为一种新的药物。
Hergenrother说,"这些革兰氏阴性菌具有外膜,这种外膜是抗生素或潜在的抗生素不能够穿透的。抵抗它们的任何一种药物几乎总是通过一种特殊的被称作孔蛋白的通道进入的。这种孔蛋白提供它们存活所需的氨基酸和其他的化合物。"
Hergenrother团队着重关注他们自己构建的复杂的分子数据库,而没有使用商业的化学物数据库。这些复杂的分子是植物和微生物的天然产物,也是他们在实验室中进行修饰的对象。
Hergenrother说,"几年前,我们已发现通过一系列有机化学反应步骤,我们能够将天然产物转化为与这些母体化合物看起来非常不同的分子。"他说,这些新的分子要比大多数商业上获得的化学物种类更具多样性。Hergenrother团队利用这种方法获得了600多种新的化合物。
Hergenrother团队单个地测试这些化合物的抗革兰氏阴性菌活性,寻找那些成功地在这些细菌细胞内部聚集的化合物。
Hergenrother说,"找到的几种化合物都具有氨基基团,因此,我们开始以此为基础进一步开展实验。"
Hergenrother团队测试了更多的具有氨基的化合物,结果他们的成功率也增加了。但是这并不是进入革兰氏阴性菌细胞内部所需的唯一特征。Hergenrother说,"具有氨基是必需的,但并不是足够的。"
利用一种计算方法,Hergenrother团队发现了穿透所需的三个关键特征:为了进入革兰氏阴性菌细胞内部,一种化合物必须具有不受其他的分子组分阻碍的氨基;它必须是非常刚性的(柔软的化合物更可能停留在孔蛋白中);它必须具有"较低的球形结构",更简单地说,它必须是平直的。
为了测试这些指导方针,Hergenrother团队给脱氧尼博霉素(deoxynybomycin, DNM)添加氨基。脱氧尼博霉素是20世纪60年代由当时的伊利诺伊大学化学教授Kenneth Rinehart Jr.开发出来的。他们选择这种化合物是因为它是一种强效地杀死革兰氏阳性菌的分子,具有其他的优良特征:刚性和较低的球形结构。通过将氨基添加到这个分子的合适位置上,他们将DNM转化为一种广谱抗生素,他们称之为6DNM-amine。
发现穿透革兰氏阴性菌外膜的化合物是比较重要的,但是抗生素也必须杀死这些细菌。Hergenrother说,之前的研究已提示着在200种随机选择的穿透革兰氏阴性菌细胞的化合物中,大约仅有一种化合物也可能杀死这些细菌。
他说,"这种几率还是不错的,总比50万种化合物当中没有一种化合物能够杀死这些细菌要好。"
抗生素耐药性越来越成为威胁全球人口健康的一大威胁,2014年英国首相牵头的一项研究就预测道,如果抗生素耐药性问题没有被有效遏制的话,在不到35年的时间里将会有更多人死于抗生素耐药性的菌株感染,而这要比癌症死亡更可怕,因此对于研究人员而言,开发新型抗生素来阻断耐药菌株的感染就显得尤为重要了。
日前,一项刊登在国际杂志Structure上的研究报告中,来自麦吉尔大学的研究人员通过研究在原子细节上阐明了细菌的特殊激酶如何介导对大环内酯类抗生素产生耐药性,这类抗生素是一类广泛使用的抗生素,其能够用来进行青霉素过敏症患者的治疗,这项研究也首次阐明了细菌激酶识别并且化学性破坏大环内酯类抗生素的机制。
此前研究人员熟知这种激酶,同时研究者发现,这种激酶在化学和结构层次发挥作用并不是一个简单的过程,2009年,研究者Albert Berghuis开始对这种激酶进行大量深入研究,在进行前期的准备后,下一步研究人员将会制造该酶类的结晶体(类似于糖晶体),随后他们再利用X射线对这些晶体进行照射。研究人员制造这些警惕并且对相关数据进行分析又花费了三年时间,最终他们清楚呈现了原子水平下的激酶图像,同时阐明了这些激酶同不同大环内酯内抗生素结合的机制。
研究人员发现,这些激酶有着强大的能力能够介导细菌对多种不同的大环内酯类抗生素产生耐受性,其中两种介导对抗生素产生耐药性的酶类是目前正在使用的治疗性抗生素;研究者Berghuis解释道,目前我们能够清楚阐明超级细菌如何利用这种关键酶类来介导对大环内酯类抗生素产生耐药性,这就能够帮助我们对这些抗生素进行一些小的修饰从而使得这些酶类不再同这些药物相互作用,当然这对于后期开发新一代不易让细菌产生耐受性的抗生素也提供了新的线索。
后期研究人员将会开发新型经过改善修饰的大环内酯类抗生素,并且对这些抗生素进行检测,这也是目前唯一能够抵御超级细菌传播的方法;抗生素耐药性是一个多方面的问题,研究者的研究仅仅是其中一个方面,他们还应该考虑其它方面,比如如何限制抗生素的过度使用,最终当一种全方位多管齐下的策略被开发的时候,研究人员或许就能够成功解决威胁全球人类健康的耐药细菌了。
当你体内还有个小人时,你总是对你吃的东西格外留意。因此,担心服用抗生素是合理的,特别是当常常读到"抗生素会增加流产风险"的新闻的时候。但是您知道什么比增加流产风险更糟糕么?那就是细菌感染。常常会有新闻告诉我们,怀孕期间服用抗生素不合适,会增加流产的风险。关于这个问题,科学家早已知道抗生素会增加流产的风险,但是有时候有些风险必须得承受。
这个新的研究在《 Canadian Medical Association Journal 》发表,作者们指出,妊娠早期给予了特别的抗生素,与流产的风险增加有关。在服用抗生素的孕妇中,有16.4%流产。而没有服用抗生素的妇女中,有12.6%仍然流产。这种几个百分点增长并不是微不足道的,我们不应该忽略小的差异。 但是如果没有发明抗生素,那么这个流产的比例会更大。
细菌感染对胎儿来说真是个坏消息。他们没有成熟的免疫系统,细菌可能会对生长中的宝宝造成破坏,即使细菌不穿越胎盘。研究抗生素风险的麻烦是,将药物的危险与感染危险区别开来非常困难。您需要抗生素的事实,意味着您可能更有可能导致流产的并发症。这就是为什么这项新研究的作者,特别说他们不能排除感染的影响。这真的是问题的关键:你实际上无法控制感染的影响,因为为健康的孕妇开一种抗生素是不负责任的。
如果你怀孕了,而且你得到甲硝唑的处方,你已经有更多的并发症的危险,因为你已经有了细菌感染。也许这是细菌性阴道炎简单感染。如果在孕期感染,容易造成婴儿早产,甚至伴随着新生儿脑出血和听力损伤。
好消息是,许多常见的抗生素,在怀孕期间服用被认为是完全安全的。青霉素(阿莫西林)不会对妈妈或婴儿造成伤害,只要它们不过敏,这通常是针对普通细菌感染的药物。如果母亲感染了其他细菌,无法使用常见的抗生素治疗,那么很难说抗生素会导致流产概率增加,因为即使不使用抗生素,因为感染细菌的原因,流产的概率一样会明显增加。
最近发表在《gut》杂志上的一篇文章指出:早年间持续性地使用抗生素将会导致大肠息肉的增生,对对于癌症的发生具有潜在的威胁。这一发现与许多相关的研究共同表明,肠道的微生物群组对于癌症的发生具有至关重要的作用。
此前的研究表明,使用抗生素会导致肠道癌症的发生,但这些研究持续时间都较短,因此这一相关性的强度饱受质疑。另外,抗生素与肠息肉的增生之间的关系也并不清楚。
为了搞清楚这些问题,研究者们通过分析此前的调查数据(该数据包含30到55岁之间的121700名护士。这些参与者们被要求填写了包括生活习惯、用药史、病史等在内的一系列健康问题)。
在这项研究中,研究者们分析了其中16642名60岁以上的护士群体。这些参与者能够提供从20岁到59岁之间的抗生素用药史以及肠息肉增生的病史。在这一时期,有1195名参与者被检查出患有腺瘤。
研究结果表明,最近一段时间内的抗生素用药水平与腺瘤发病的风险之间并没有明显的相关性,但长期的抗生素使用历史则与腺瘤的发生有较强的相关性。
相比20岁到30岁之间没有接触过抗生素的人群,接触过的人群患腺瘤的比例高出36%之多。相似地,40到50岁之间每两个月(或更短的时间内)使用一次抗生素的患者被诊断出患腺瘤的风险相比其他人群也要高出69%。
当然,由于该研究属于观察性的试验,因此难以得出确切的结论,但是这一相关性背后有着合理的生物学解释。抗生素的过度使用会导致肠道微生物菌群受到影响,而此前有研究表明肠道微生物结构的紊乱对于大肠癌的发生具有一定的影响。
因此,为了减小肠道癌症的发生风险,我们应当谨慎使用抗生素。
日前,一项刊登在国际杂志the American Journal of Respiratory and Critical Care Medicine上的研究报告中,来自伯明翰大学和纽卡斯尔大学的研究人员通过研究开发了一种治疗抗生素耐药性细菌感染的新型疗法。文章中,研究者发现,利用一种不寻常的方法从血液中移除抗体就能够降低细菌慢性感染的副作用、患者住院的时间以及抗生素的使用。
这项研究中,研究者鉴别出了两名遭受铜绿假单胞菌慢性感染的支气管扩张症患者,其中一名患者为64岁男性,其在十五岁时被诊断为支气管扩张症,另外一名患者未69岁女性,其儿时就患有支气管扩张症;铜绿假单胞菌对多种抗生素都有耐药性。支气管扩张症能够诱发肺部气管永久性的扩张,其仅在英国就影响着30万患者的健康,患者主要症状表现为慢性咳嗽、呼吸短促、咳血以及胸疼。
慢性的铜绿假单胞菌肺部感染常常发生于支气管扩张症患者机体中,研究者Ian Henderson解释道,这些患者通常血液中都含有过量特殊的抗体,相比正常状况下抗体相关的保护效应而言,在这些患者机体中,这些抗体会阻断机体免疫系统杀灭铜绿假单胞菌的能力,同时还会使得患者机体肺部的疾病恶化,因此研究人员决定通过研究去除患者机体中的这些特殊的抗体,来观察患者后期的表现情况。
研究者认为,他们需要一种新方法来解决支气管扩张症患者的健康问题,随后研究人员同从事肾脏和免疫学研究的科学家们进行合作,利用血浆去除术来进行后期更为深入的研究,血浆去除术包括从循环系统中移除、治疗并且返回等过程,患者每周要进行5次治疗来移除血液中的特殊抗体,随后研究者从献血者机体中也移除了这些抗体,这种新型疗法就能够帮助患者机体恢复杀灭铜绿假单胞菌的能力。
研究者Henderson表示,所有的患者都能够快速恢复机体健康,而且能够更加独立地完成一些事情,同时其活动能力也得到了改善。基于本文研究,研究人员就能够利用这种新方法来明显改善患者的生存状况,下一步研究者将会进行更长期的研究来调查是否早期干预,同时进行并不激进的疗法能够帮助抑制患者疾病的进展。
最后研究者说道,这项研究中我们首次描述了对细菌感染性疾病的抗体依赖性增强疗法,当然其或许能够应用到其它细菌感染的治疗中,也有望作为新型疗法来治疗某些抗生素耐药性的感染。
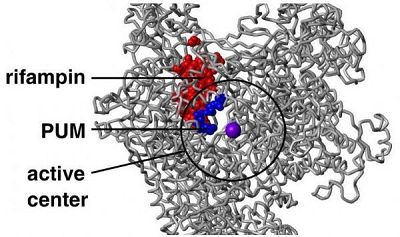
近日,一项刊登在国际杂志Cell上的研究报告中,来自美国罗格斯大学等机构的研究人员通过研究发现了一种能够有效抵御耐药性细菌的新型抗生素-pseudouridimycin,这种抗生素由来自土壤样本中的微生物所产生,通过在测试管中进行试验,这种新型抗生素能够杀灭一系列药物敏感性和耐受性的细菌。
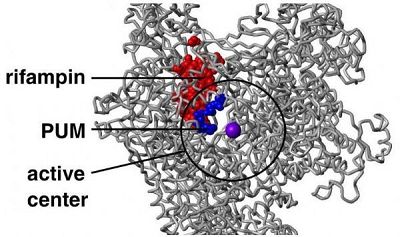
文章中,研究人员报道了这种新型抗生素pseudouridimycin的作用及机制;该抗生素能够通过一种结合位点来抑制细菌细胞中RNA聚合酶的功能,但其作用机制并不同于当前所使用的抗生素-利福平;因为pseudouridimycin能够通过一种不同与利福平的结合位点来抑制细菌生长,因此该抗生素往往不会促进细菌产生与利福平的交叉耐药性。
抗生素pseudouridimycin起着细菌RNA聚合酶核苷类似物抑制剂的作用,也就意味着其能够模仿三磷酸核苷(NTP),而NTP是细菌RNA聚合酶用来和成RNA的基本结构原件,这种新型抗生素能够通过占领NTP结合位点同细菌RNA聚合酶上的该位点紧密结合,从而抑制NTPs的结合。该抗生素是首个核苷类似物抑制剂,其能够选择性地抑制细菌RNA聚合酶的功能,但对人类机体RNA聚合酶并无影响。
研究者Richard H. Ebright说道,因为细菌RNA聚合酶的NTP结合位点几乎拥有和人类RNA聚合酶相同的结构和序列,很多研究人员都认为这不可能作为核苷类似物抑制剂抑制细菌RNA聚合酶的结合位点(并非是人类细胞的RNA聚合酶);但研究者发现,抗生素pseudouridimycin含有一种侧链,其能够到达NTP结合位点的外部,并且同细菌RNA聚合酶中存在的临近位点相结合,相比人类RNA聚合酶而言,最终新型抗生素就能同细菌RNA聚合酶紧密结合。实际上,pseudouridimycin作为一种核苷类似物抑制剂还能够帮助解释该化合物引发的耐药率较低的原因。
这种新型抗生素能够同NTP结合位点的必要残基相互作用,在没有失去RNA聚合酶活力和细菌活性时NTP结合位点并不会发生改变,对NTP结合位点的改变就能够干扰其同新型抗生素的结合,从而干扰RNA聚合酶的活力,诱发细菌死亡,但并不会引起细菌产生耐药性。研究者Stefano Donadio表示,选择性抑制病毒核苷酸聚合酶的核苷类似物抑制剂或许还会对HIV和丙肝的治疗带来突破性的变革,抗艾滋病的药物齐多夫定、扎昔他宾等都是核苷类似物抑制剂,丙肝药物索非布韦等也是此类抑制剂。
最后研究者表示,本文研究发现阐明了利用天然化合物来治疗疾病的重要性,微生物具有数亿万年的历史,其往往能够产生有效杀灭其它微生物的特殊"化学武器",因此深入探究微生物所产生的天然化合物或许也是未来开发治疗耐药性细菌感染的一个新方向。
在一项新的研究中,来自瑞典、法国、比利时和瑞士的一个研究团队发现一种方法逆转对一种用于治疗肺结核的抗生素药物产生的耐药性。在他们发表在2017年3月17日的Science期刊上的论文中,该团队描述了他们如何筛选激活用于活化ethionaide的不同通路的化合物。Ethionaide是一种被用来治疗肺结核的前体分子,在体内经过代谢后产生一种真正有疗效的药物。
开发治疗细菌性感染的抗生素已明显地让这个世界变得更加健康。不幸的是,随着时间的推移,细菌不断地进化出挫败这些抗生素的能力,从而让我们再次处于危险当中。也正因如此,科学家们一直在寻找新的疗法,或者在一些情形下,利用新的技术让旧的疗法再次起作用。在这项新的研究中,这些研究人员发现一种方法让ethionaide再次有效地治疗被耐药性结核分枝杆菌(Mycobacterium tuberculosis)菌株感染的病人。
在1950年代晚期,ethionaid是作为一种肺结核治疗药物而被开发出来的。它被结核分枝杆菌中发现的酶EthA激活。一经激活,ethionaid就会攻击这种细菌。随着时间的推移,很多结核分枝杆菌菌株通过发生不再激活这种前体分子的EthA突变而对ethionaide产生耐药性,从而使得它不再适合用于治疗。为了绕过这个问题,这些研究人员在经过一番搜寻后,发现一种被称作SMARt-420的原型分子通过采取一种不同的途径(与第二个被称作EthR2的基因相互作用)激活ethionaide。他们发现在服用一剂ethionaide后,给病人一剂这个小分子会恢复ethionaide破坏一系列结核分枝杆菌菌株的能力:测试结果表明仅在三周后,它就降低在病人肺部中发现的细菌载量,它的效果类似于在耐药性产生之前,ethionaide独自抵抗结核分枝杆菌的疗效。
这些研究人员当前正与葛兰素史克公司(GlaxoSmithKline)和生物技术公司Bioversys合作进一步将这个原型分子开发为一种能够大规模制造和销售的药物。他们也正在探究是否能够利用这种相同的技术或一种类似的技术战胜其他的细菌产生的耐药性。